Explainable AI in Drug Design: Perturbation based molecular attributions using Graph Convolutional Networks
This is the thesis project I have done to graduate at the Master's Degree in Artificial Intelligence and Robotics under the supervision of prof. Roberto Capobianco.
Abstract
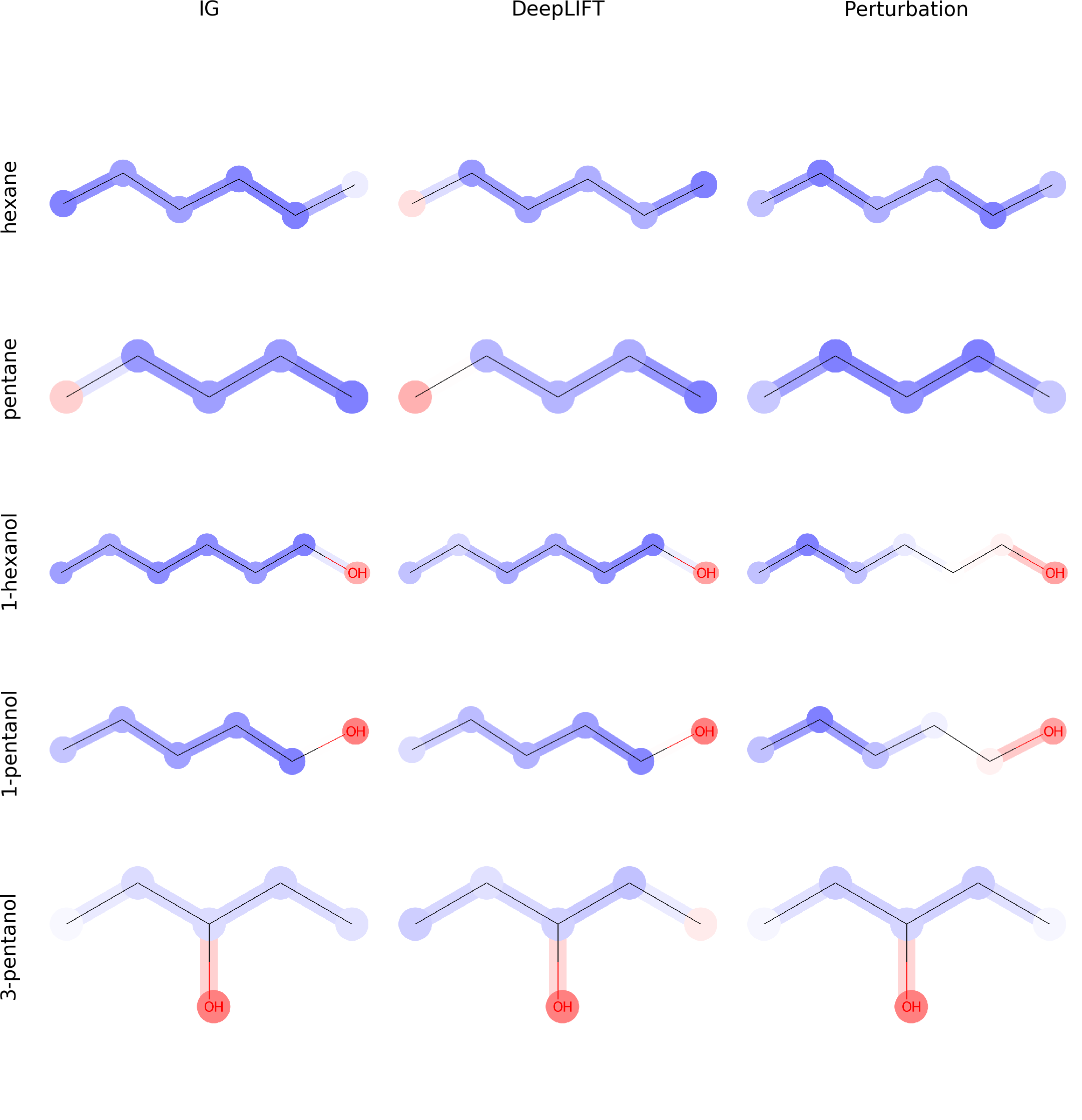
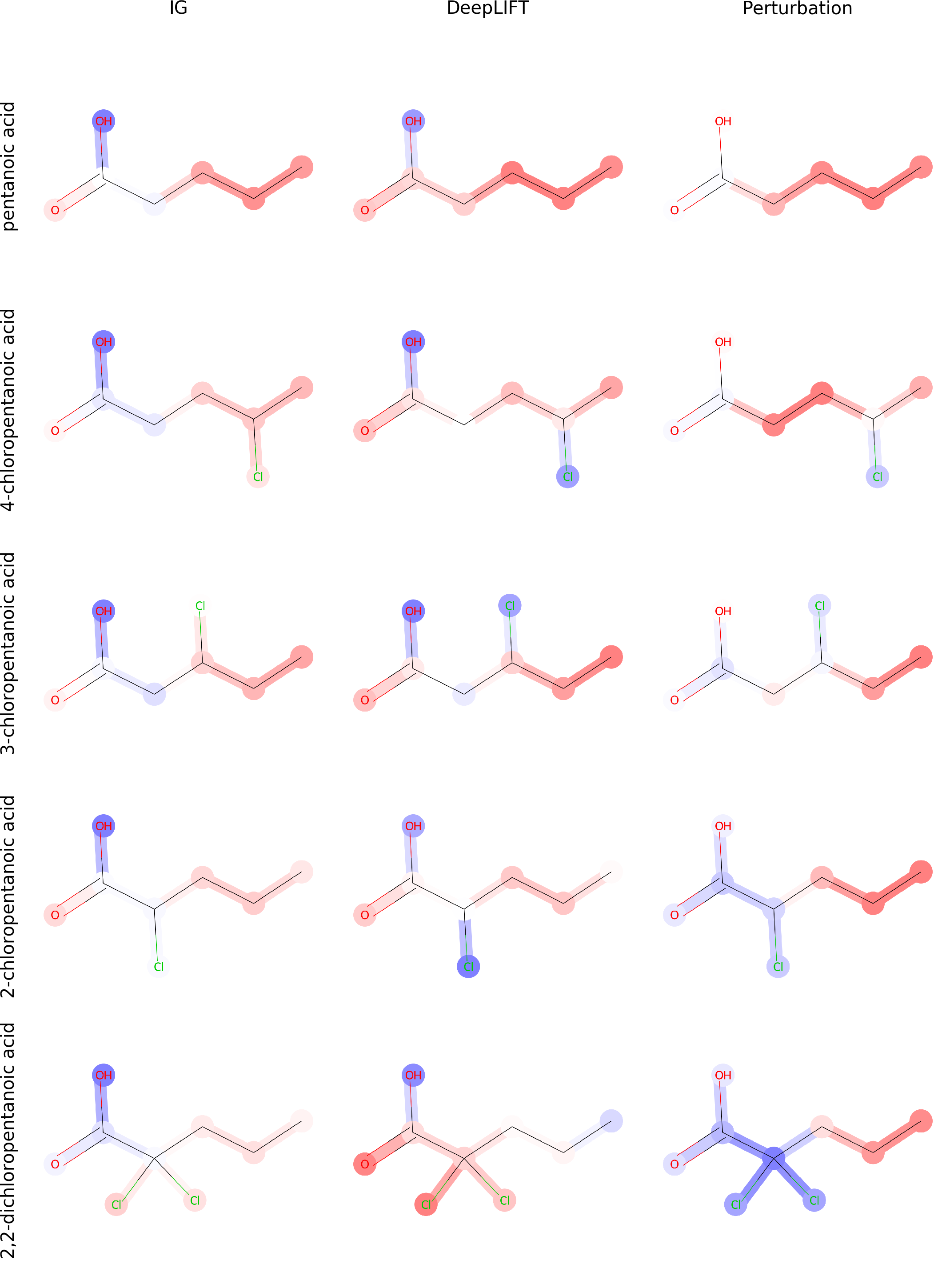
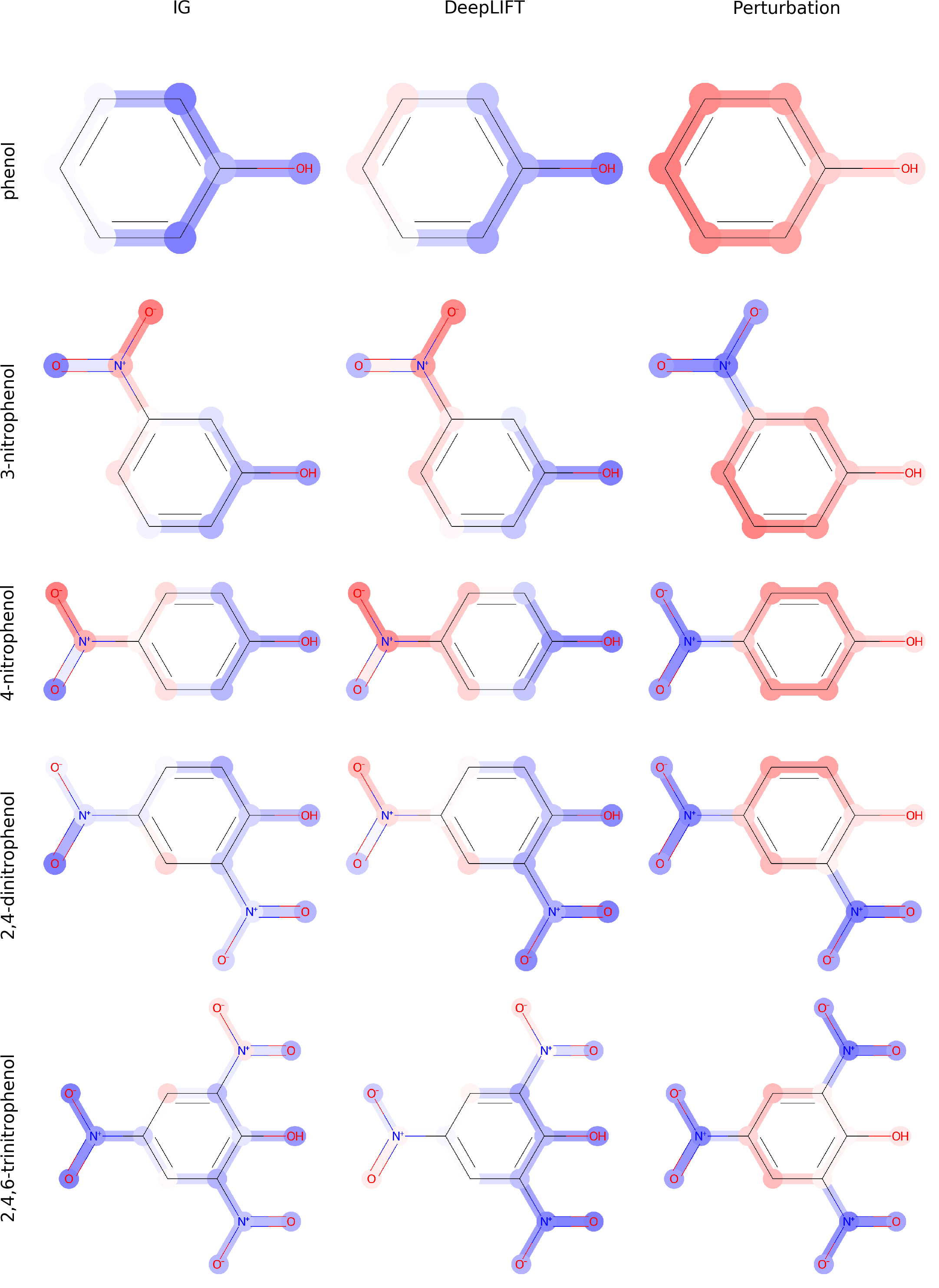
Since the introduction of artificial intelligence in medicinal chemistry, the necessity has emerged to analyse how molecular property variation is modulated by either single atoms or chemical groups. In this paper, we propose to train graph-to-graph neural network using semi-supervised learning for attributing structure-property relationships. As initial case studies we apply the method to solubility and molecular acidity while checking its consistency in comparison with known experimental chemical data. As final goal, our approach could represent a valuable tool to deal with problems such as activity cliffs, lead optimization and de-novo drug design.